5.3 EMP_feature_convert
In studies, the naming of genes varies depending on the database used. Therefore, for transcriptomic enrichment analysis, gene names need to be converted to their corresponding unique ID (often called ENTREZID). In metabolomics analysis, there is also the problem of ID conversion among multiple databases. Module EMP_feature_convert provides easy conversion of gene ID and metabolite ID between multiple databases.
5.3.1 Conversion for Gene ID
🏷️Example:
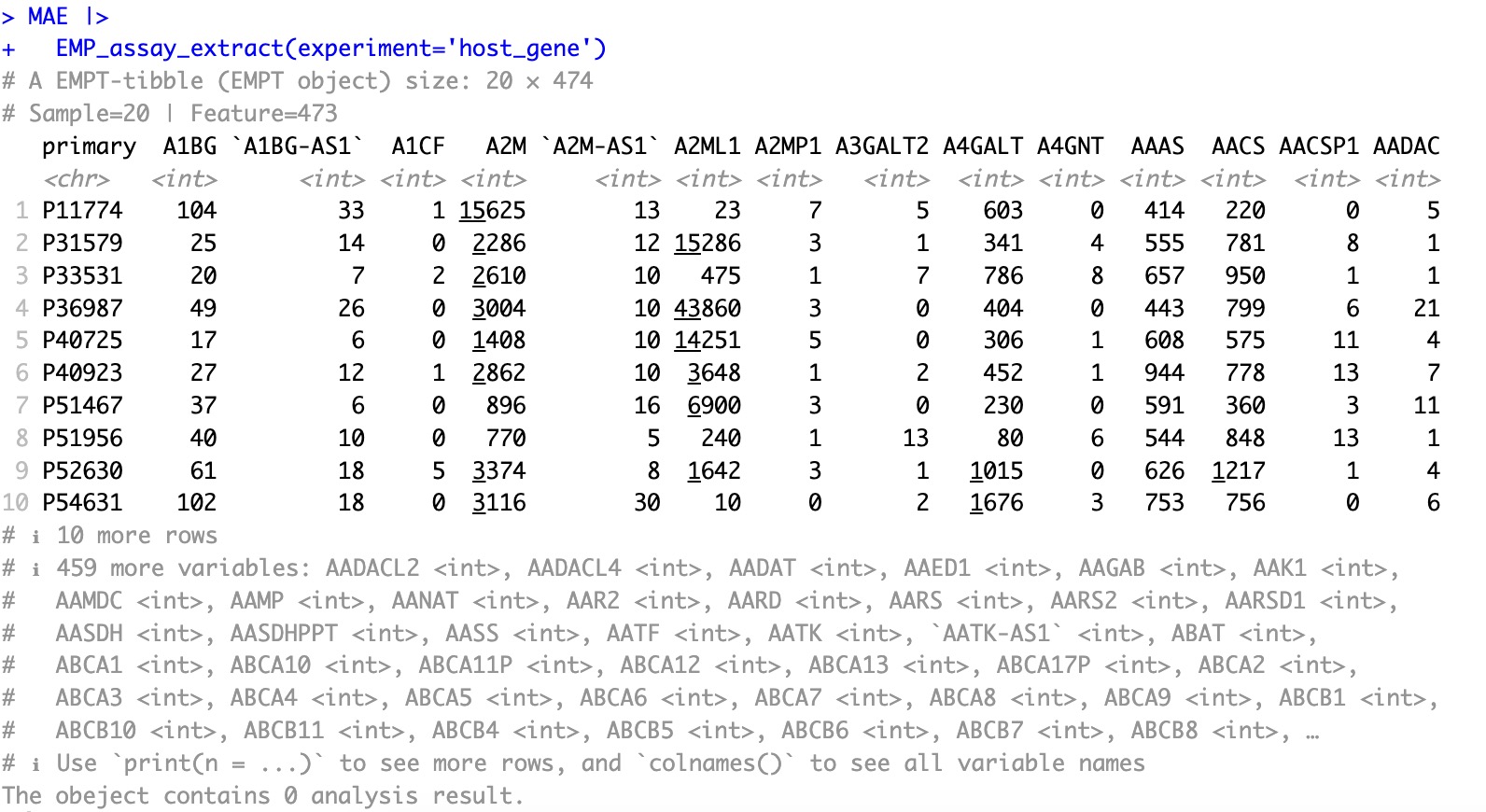
Before conversion:
MAE |>
EMP_assay_extract(experiment='host_gene')
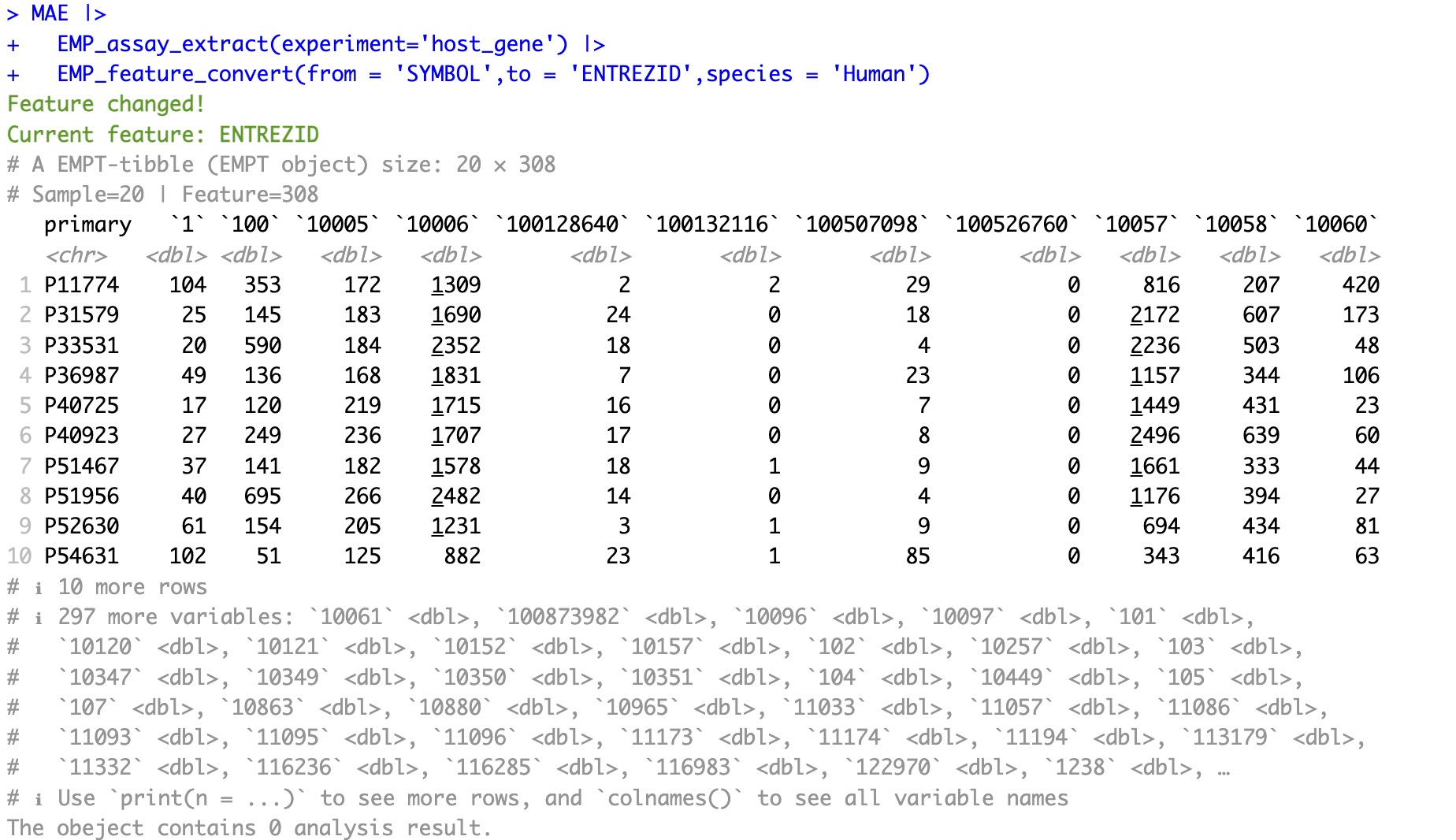
After conversion:
This package includes reference gene sets for four commonly used species:
Human, Mouse, Pig, and Zebrafish. If your data pertains to a different species, you can specify the corresponding reference dataset using the parameter OrgDb. Click here for more species details.
MAE |>
EMP_assay_extract(experiment='host_gene') |>
EMP_feature_convert(from = 'SYMBOL',to = 'ENTREZID',species = 'Human')
5.3.2 Conversion for Metabolite ID
🏷️Example:
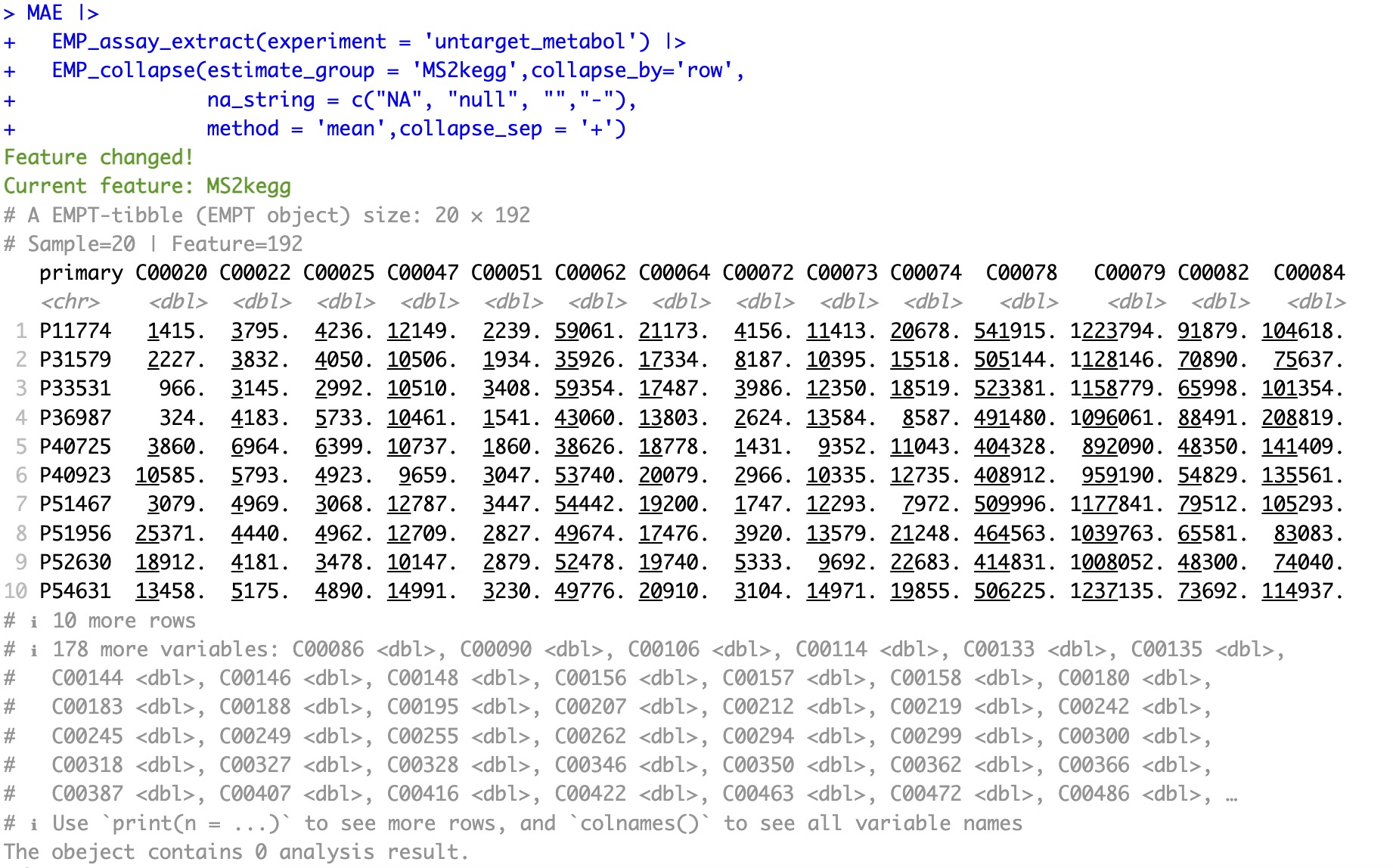
Before conversion:
MAE |>
EMP_assay_extract(experiment = 'untarget_metabol')|>
EMP_collapse(na_string=c('NA','null','','-'),
estimate_group = 'MS2kegg',method = 'sum',collapse_by = 'row')
🏷️After conversion:
Convert metabolite from Compound annotation of KEGG to ID annotation of Human Metabolome Database.
MAE |>
EMP_assay_extract(experiment = 'untarget_metabol'),
EMP_collapse(na_string=c('NA','null','','-'),
estimate_group = 'MS2kegg',method = 'sum',collapse_by = 'row') |>
EMP_feature_convert(from = 'KEGG',to='HMDB')

5.3.3 Conversion for Microbial Taxonomy
Microbial data are generally divided into multi-level annotations such as Kingdoms, Order, Family, Genus, and Species for downstream data analysis. However, due to the existence of repeated annotations in some rare taxa, such as the situation where the genus-level annotations of some bacteria are consistent but the family-level annotations are inconsistent, this function can complete the annotation of microbial data at each level. (Details are explained in Chapter 10.4)
🏷️Example:
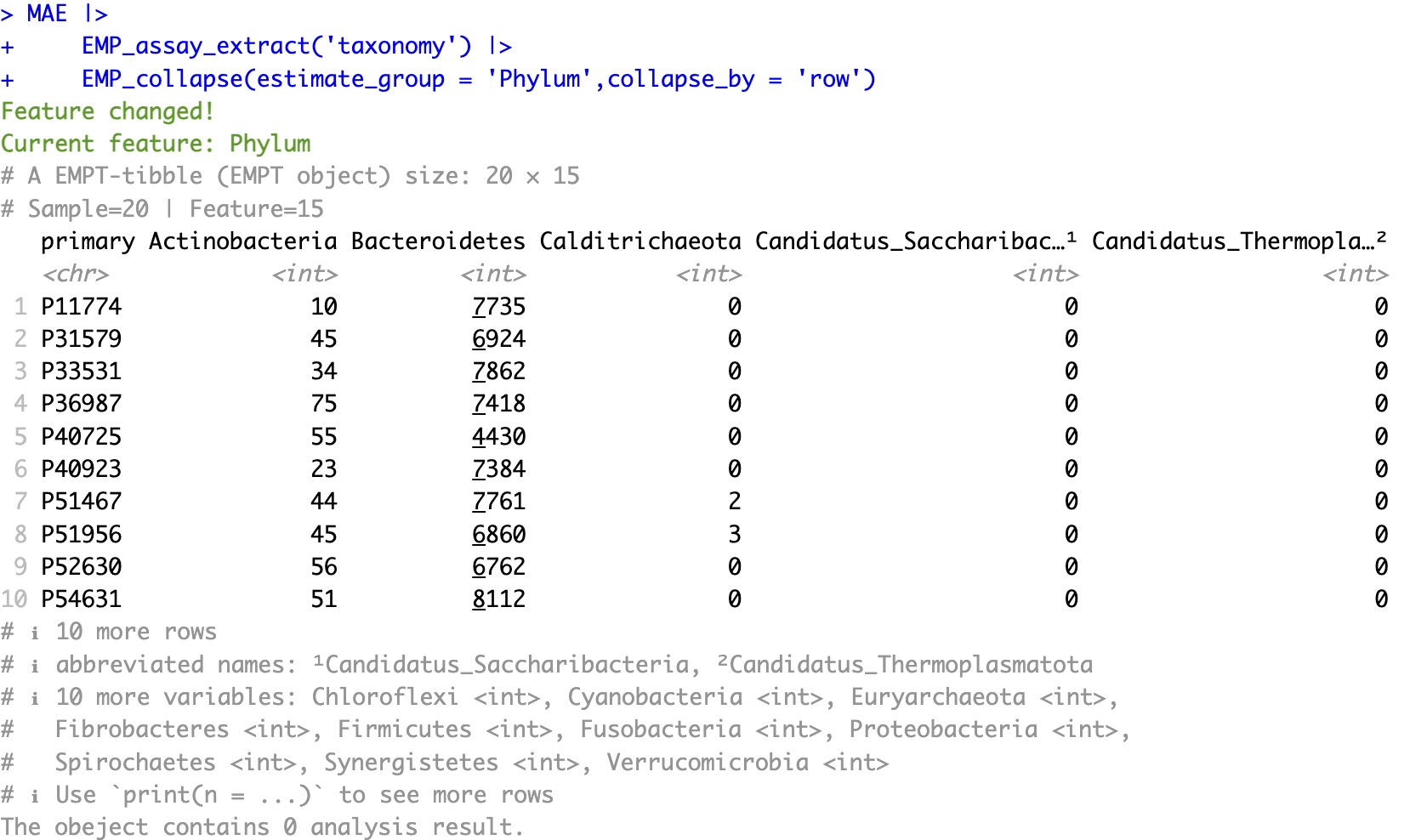
Before conversion:
MAE |>
EMP_assay_extract('taxonomy') |>
EMP_collapse(estimate_group = 'Phylum',collapse_by = 'row')
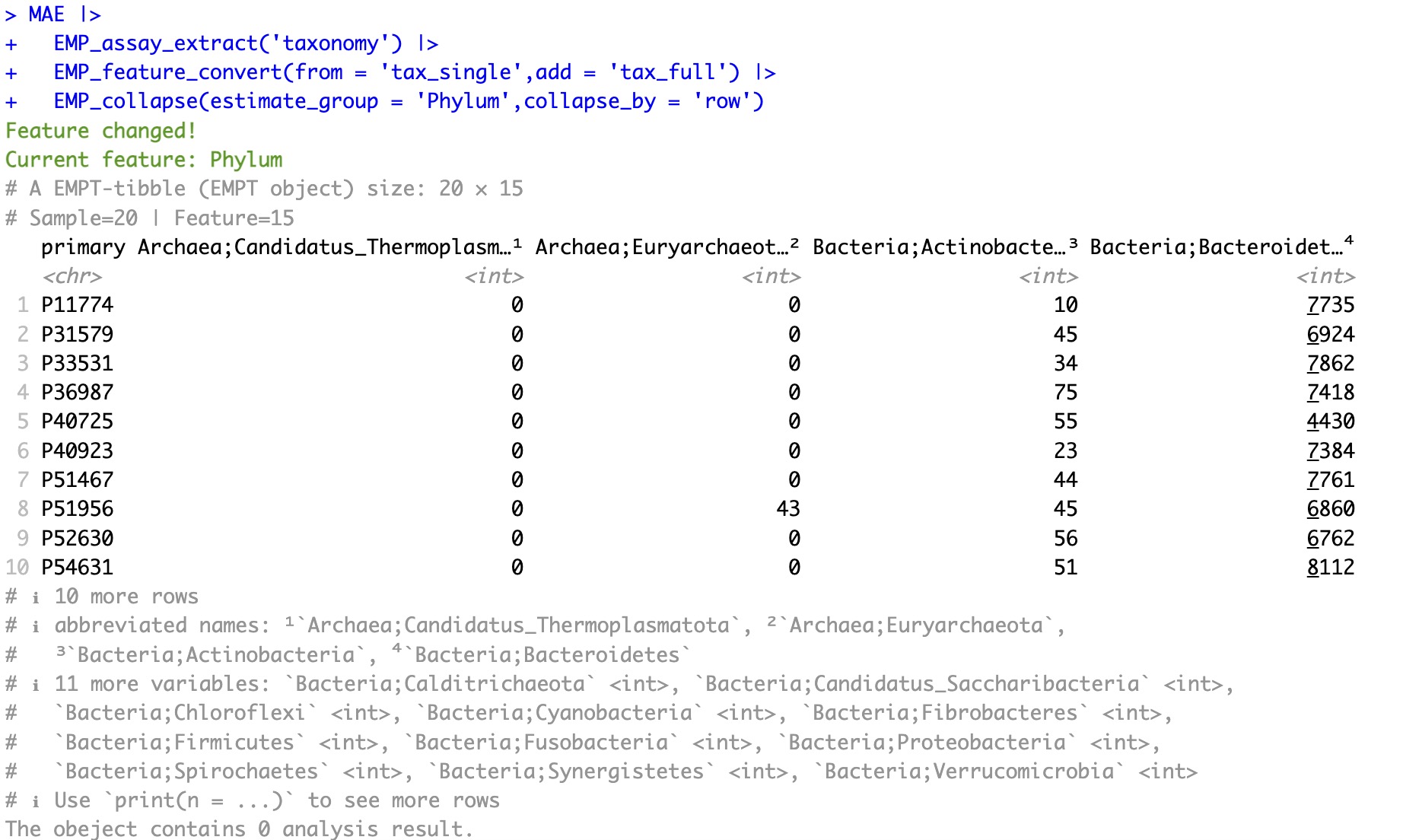
After conversion:
This function can only work before collpsing microbial data based on taxonomy profile.
MAE |>
EMP_assay_extract('taxonomy') |>
EMP_feature_convert(from = 'tax_single',add = 'tax_full') |>
EMP_collapse(estimate_group = 'Phylum',collapse_by = 'row')
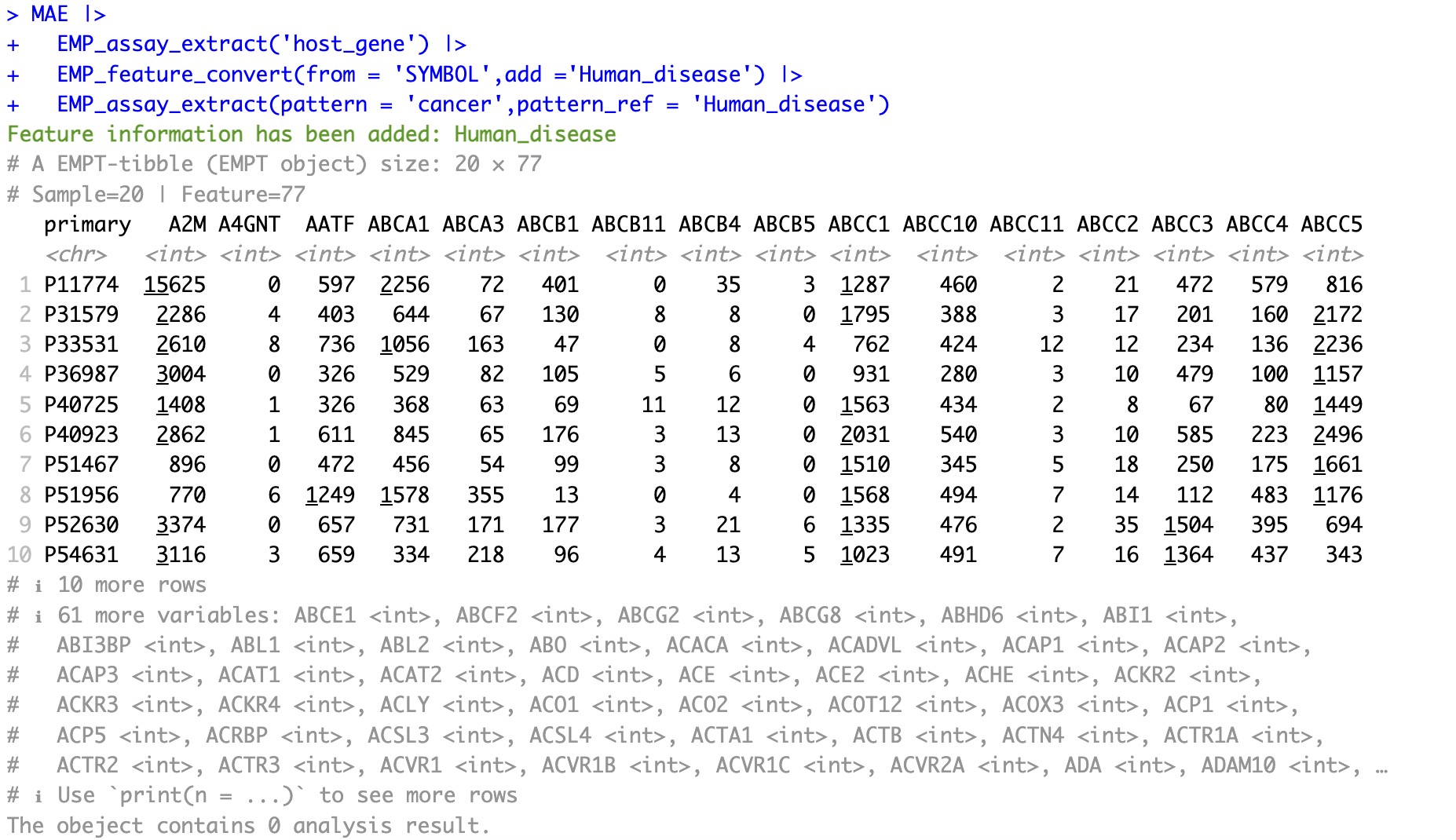
5.3.4 Add feature-related disease annotations
This module supports SYMBOL, ENTREZID, ko, and ec.
🏷️Example: Add human disease annotations and screen out genes related to cancer
MAE |>
EMP_assay_extract('host_gene') |>
EMP_feature_convert(from = 'SYMBOL',add ='Human_disease') |>
EMP_assay_extract(pattern = 'cancer',pattern_ref = 'Human_disease')